Next-generation phenotyping integrated in a national framework for patients with ultrarare disorders improves genetic diagnostics and yields new molecular findings

Main
A recent analysis of the Orphanet database showed that around 3–6% of the global population have a rare disease (that is, a disease with a prevalence of <1 in 2,000) and that 72% of such cases may have a genetic cause1. Rare diseases thus represent a substantial global health burden. However, only a minority of patients suspected to have a rare disease receive both a definite clinical diagnosis and a confirmatory molecular test result. This concerns in particular the subset of patients with ultrarare disorders that are defined in the European Union as affecting no more than one person in 50,000 and that follow a long tail distribution with respect to their frequency (Regulation (EU) No. 536/2014). It is estimated that roughly 80% of the more than 5,000 rare genetic diseases have a prevalence below one in a million.The International Rare Disease Research Consortium therefore stated that, by 2027, all patients who come to medical attention with a suspected rare or ultrarare disease should be diagnosed within 1 year if the respective disorder has been described in the medical literature. Since many rare diseases are Mendelian in nature, comprehensive genetic testing is a key element to achieve that goal.
In Germany, around 90% of the population has statutory health insurance, and the current reimbursement scheme allows physicians to request chromosome analyses, molecular karyotyping and sequencing of single genes or gene panels. For example, high-resolution genome-wide array-based segmental aneusomy profiling detects a pathogenic aberration in around 19% of patients with developmental delay. Besides contiguous gene syndromes, most of the remaining rare disorders are monogenic and are caused by single nucleotide variants or small insertions or deletions (indels). However, single gene analyses or small gene panels are only likely to detect a pathogenic aberration if the phenotype is highly predictive of the molecular cause, for example, hemoglobinopathies.
For phenotypes with high genetic heterogeneity, such as neurodevelopmental disorders, genetic investigation is more challenging. For intellectual disability, for example, studies so far have identified disease associations for more than a thousand genes. For these disorders, research has shown that exome sequencing can be more cost-effective than sequencing potentially multiple gene panels. However, this is also accompanied by more genetic variants that have to be assessed. Therefore, a clear indication for exome sequencing and efficient data analysis strategies are crucial. Between 2018 and 2020, a novel diagnostic concept within the German healthcare system was evaluated in the prospective study TRANSLATE NAMSE.
This involved standardized structures and procedures and multidisciplinary teams (MDTs) at ten university hospital-based centers for rare diseases (CRDs). The MDTs conducted a three-step diagnostic process: (1) primary review of patient records; (2) selection of diagnostic procedures, including a possible recommendation for exome sequencing; and (3) evaluation of all findings, including genetic variants. A key goal was to investigate whether exome sequencing would facilitate the diagnosis of ultrarare disorders or even the delineation of novel monogenic disorders. In this work, we report the molecular findings of this study.
Furthermore, we investigated how phenotypic features can be used to estimate the probability that a molecular diagnosis can be established with exome sequencing (YieldPred). In a companion study, we also assessed the extent to which the results from computer-assisted pattern recognition in facial dysmorphism contribute to variant interpretation (prioritization of exome data by image analysis, PEDIA). The present analyses demonstrated that exome sequencing facilitated the diagnosis of ultrarare genetic diseases and novel gene–disease associations and that artificial intelligence (AI)-driven technologies improved the diagnostic yield for ultrarare genetic disorders.
Results
Phenotypic characteristics of the study cohort
Between 2018 and 2020, a total of 5,652 individuals (2,033 adults and 3,619 children) with a suspected rare disorder were enrolled in TRANSLATE NAMSE by CRDs at ten German university hospitals. The present analyses were performed using the data from a total of 1,577 of these 5,652 patients (268 adults, 1,309 children). In these individuals, the MDT at the respective CRD considered a genetic cause as plausible and exome sequencing as the most suitable test (exome sequencing cohort, Supplementary Table). Each of these 1,577 individuals was assigned to one of six major disease categories by the respective CRD physician=. The majority of children were assigned to the disease category ‘neurodevelopmental disorders’ (n = 702, 54%), and the largest proportion of adults were assigned to the disease category ‘neurological or neuromuscular disorders’ (n = 117, 44%). Smaller proportions of adult and pediatric cases were assigned to the groups ‘organ malformation’, ‘endocrine/metabolic disorders’, ‘immune/hematologic disorders’ and ‘cardiovascular disorders’. Patient phenotypes were also annotated with terms of the Human Phenotype Ontology (HPO) by the respective CRD physicians. On average, five HPO terms were specified per individual (Supplementary). The phenotypes within the present cohort were visualized by projecting the patient-specific HPO terms into a two-dimensional space. While most patients from the same disease group were in close proximity, the clusters showed a partial overlap. For example, many patients categorized within ‘neurological or neuromuscular disorders’ also showed HPO terms typically associated with ‘neurodevelopmental disorders’ and vice versa (Supplementary). This suggests that grouping patients into single disease groups may be overly simplistic.Fig. 1: Workflow in the TRANSLATE NAMSE project and phenotypes in which exome sequencing was performed.
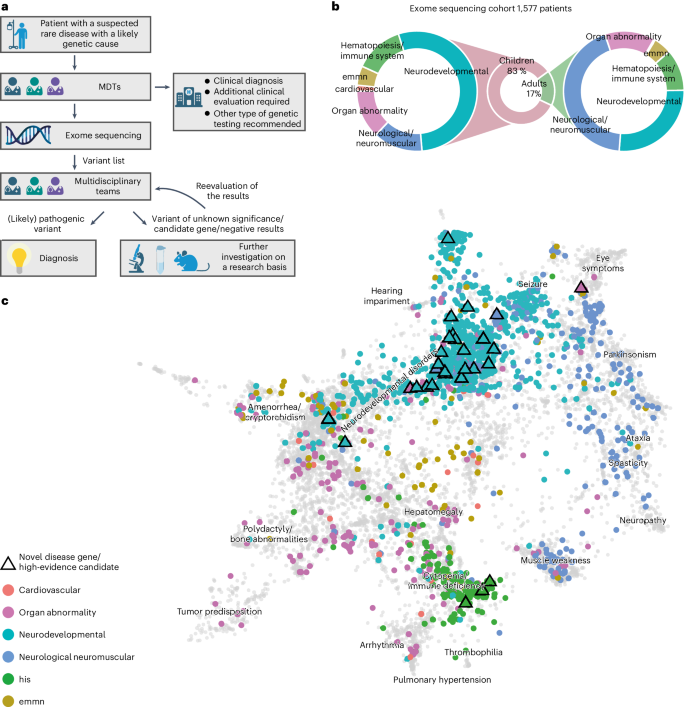
a, Patients with a suspected rare disease were referred to a MDT and deeply phenotyped using HPO terminology. If a genetic etiology was considered likely, exome sequencing was performed. The MDT then evaluated the molecular findings and could order additional analyses for variants of uncertain significance or variants in potentially novel disease candidate genes. b, Exome sequencing was performed predominantly in children. The main indications for exome sequencing in children were neurodevelopmental disorders. In adults, the main indications were neurological/neuromuscular disorders. In both children and adults, the least common disease categories were ‘cardiovascular’, ‘endocrine, metabolic, mitochondrial, nutritional’ (emmn) and ‘hematopoiesis/immune system’ (his). c, Phenotypic similarities between patients, as encoded according to their HPO terms, were visualized with UMAP. As reference, all OMIM diseases were included using their HPO annotations (gray background dots). For each patient, color coding indicates allocation to disease groups, in accordance with the leading clinical feature. An overlap is evident for patients in the neurodevelopmental and neuromuscular groups (aquamarine and blue clusters), which indicates high phenotypic similarity. This precludes the unequivocal assignment of these patients to a diagnostic group. The triangles indicate patients who contributed to the identification of a novel, high-evidence gene–phenotype association.
Diagnostic yield of exome sequencing
A molecular diagnosis was established in a total of 499 of the 1,577 patients (32%), that is, in these cases, exome sequencing identified variants that fully or partially explained the phenotype. The diagnostic yield was slightly higher in children (32%) than in adults (28%, P = 0.13, Fisher’s exact test; ) and twofold higher in patients assigned to the category ‘neurodevelopmental disorder’ than for all other disease categories (42% versus 22%, P < 0.001, Fisher’s exact test with Bonferroni correction; for single comparisons between disorder groups). Furthermore, exome sequencing found variants of uncertain significance. Specifically, these variants were enriched for missense variants (80% versus 45%, P < 0.001; Supplementary), due to lower support for pathogenicity according to the guidelines of the American College of Medical Genetics (ACMG) and the Association for Molecular Pathologists for interpretation of sequence variants.
Fig. 2: Diagnostic yield of exome sequencing depends on age and disease group.
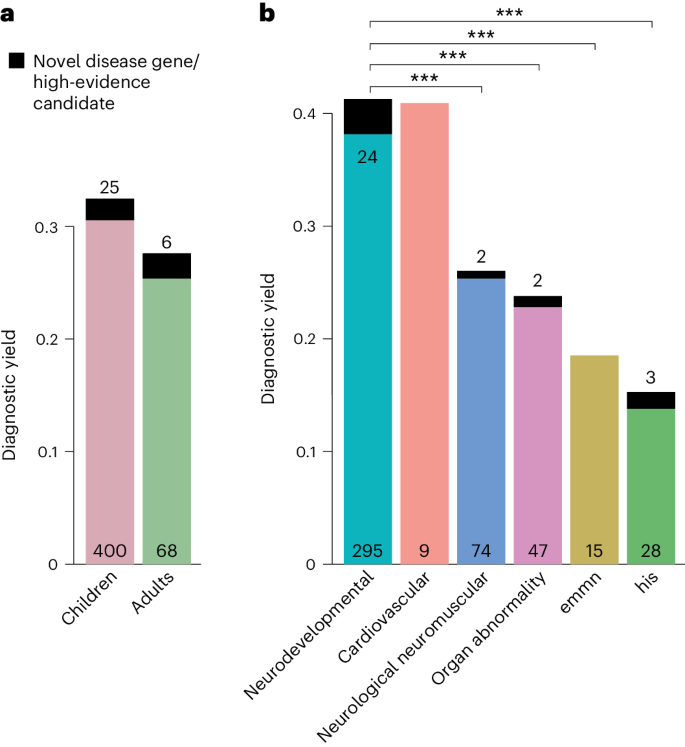
a,b, The diagnostic yield differed according to age group (adult/child) (a) and disease category (b). For all disease categories, with the exception of cardiovascular, the diagnostic yield was increased by novel DGGs and high-evidence candidate genes (dark-colored tip of the bar). The absolute number of solved cases in which a variant was found in an established disease gene is given at the bottom of each bar, and the number of solved cases attributable to a novel DGG or high-evidence candidate gene is given at the top of each bar. The entire TRANSLATE NAMSE exome sequencing cohort was considered for a and b (n = 1,577). Diagnostic yield between disease categories were compared using two-sided Fisher’s exact test. P values were adjusted by Bonferroni correction. ***P < 0.001; exact corrected P values: neurodevelopmental (ndd) versus neurologic neuromuscular P = 5.4 × 10−5, ndd versus organ abnormality P = 5.2 × 10−5, ndd versus emmn P = 5.9 × 10−4, ndd versus his P = 1.1 × 10−11. emnn, endocrine, metabolic, mitochondrial, nutritional; his, hematopoiesis/immune system.
International Conference on Genetics and Genomics of Diseases
Visit: genetics-conferences.healthcarek.com
Award Nomination: x-i.me/gennom1
Award registration: x-i.me/genreg2
Member Nomination: x-i.me/genmember
Member Registration: x-i.me/genreg1
For Enquiries: genetics@healthcarek.com
Get Connected Here
---------------------------------
---------------------------------
Pinterest: x-i.me/genpt
Twitter: x-i.me/gentw
Facebook: x-i.me/genfb
Instagram: x-i.me/genin
Youtube: x-i.me/genyt
Blogger: x-i.me/genbl
Visit: genetics-conferences.healthcarek.com
Award Nomination: x-i.me/gennom1
Award registration: x-i.me/genreg2
Member Nomination: x-i.me/genmember
Member Registration: x-i.me/genreg1
For Enquiries: genetics@healthcarek.com
Get Connected Here
---------------------------------
---------------------------------
Pinterest: x-i.me/genpt
Twitter: x-i.me/gentw
Facebook: x-i.me/genfb
Instagram: x-i.me/genin
Youtube: x-i.me/genyt
Blogger: x-i.me/genbl
Comments
Post a Comment